
2023-2024 Mount Sinai Science & Medicine Magazine
How Mount Sinai Integrates Cutting-Edge Technology With Patient-Centered Expertise

Mount Sinai Health System Names Brendan Carr, MD, MA, MS, as Next Chief Executive Officer
Nationally recognized leader in academic medicine, delivering high-quality health care as a physician, health policy researcher, and educator

Digital Patient Tools and Resources
See how our expert care is always there

Unsure What to Do When You're Not Feeling Well?
Take our symptoms assessment and view your care options with our new Check Symptoms & Get Care tool
MyMountSinai® App’s Virtual Urgent Care
Share your symptoms on the MyMountSinai® app and get answers from doctors you can trust with virtual urgent care
Founded in 1820 and now one of the world’s leading facilities for treating diseases of the eyes, ears, nose, and throat, New York Eye and Ear Infirmary of Mount Sinai (NYEE) combines a rich history of specialty care with award-winning clinical excellence in order to serve our community, as well as patients from the Tri-State area, across the nation, and internationally. In addition to our main campus near Union Square, NYEE is growing a network of satellite offices and freestanding ambulatory surgery centers, making access to the newest diagnostic services, advanced treatments, and clinical trials more convenient to our patients’ workplaces and homes.
In addition to providing skilled primary through tertiary care, NYEE is home to breakthrough clinical research that has introduced now widely practiced diagnostic and surgical techniques. Our physicians are also passing the tradition of excellence onto future generations through highly competitive residency programs in ophthalmology and otolaryngology, plus dozens of post-graduate ophthalmology fellowship positions. Through this combination of superb patient care, innovative research, and rigorous education, NYEE is maintaining a leadership position in the fields of ophthalmology, otolaryngology, head and neck surgery, and plastic and reconstructive surgery.
Mount Sinai Ophthalmologists Develop Novel Protocol to Rapidly Diagnose and Treat Eye Stroke
Feb 13, 2024 View All NewsPatients With Specific Types of Lipids May Be at Higher Risk of Developing Blinding Eye Disease
Jun 13, 2023 View All News
New Guidelines Improve Care and Practice Standards for Adults With Hearing Loss
Mar 03, 2023 View All News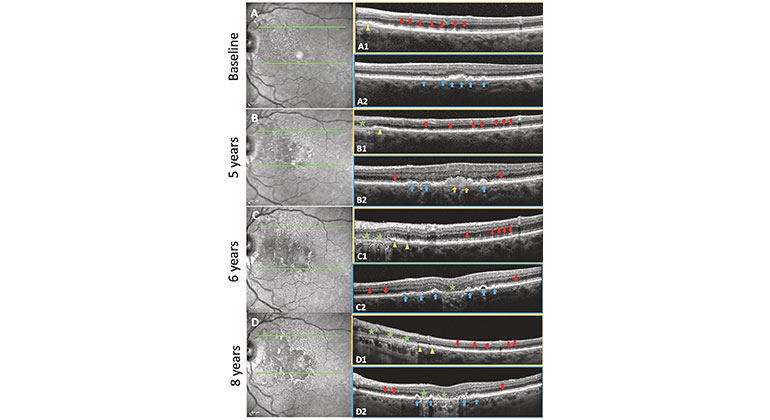

High-Tech Imaging Offers New Way to Detect Signs of Early Glaucoma
Aug 02, 2022 View All News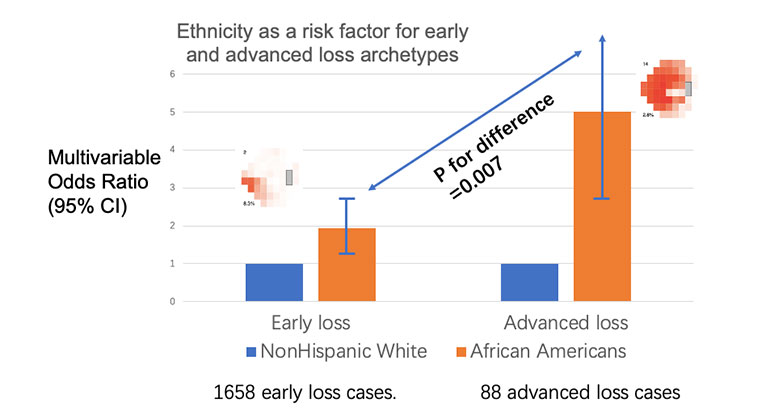
Blinding Eye Disease Is Strongly Associated With Heart Disease and Stroke
Jul 12, 2022 View All News